Any structural or functional abnormality of the ventricular myocardium, except congenital developmental defects; valvular disease; systemic or pulmonary vascular disease; isolated pericardial, nodal, or conduction system disease; or epicardial coronary artery disease, unless chronic diffuse myocardial dysfunction is present
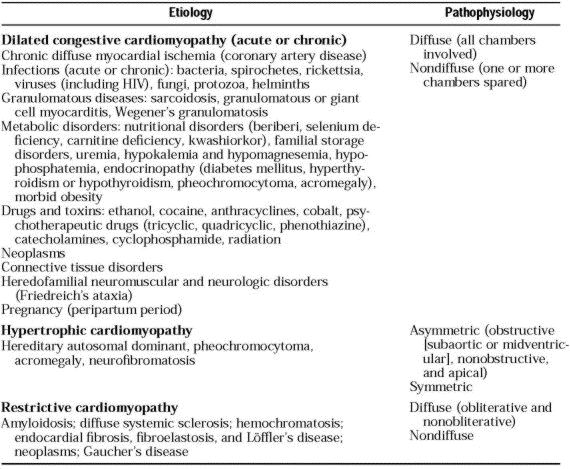
Cardiomyopathy has many causes Pathophysiologic classification (dilated congestive, hypertrophic, or restrictive cardiomyopathy) by means of history, physical examination, and invasive or noninvasive testing is most useful initially.If no cause can be found, cardiomyopathy is considered primary or idiopathic.
DILATED CONGESTIVE CARDIOMYOPATHY
Disorders of myocardial function with heart failure in which ventricular dilation and systolic dysfunction predominate.
Etiology and Pathophysiology
The most common identifiable cause in temperate zones is diffuse coronary artery disease with diffuse ischemic myopathy; for other causes.
Most commonly, at presentation there is chronic myocardial fibrosis with diffuse loss of myocytes. In some patients, the underlying pathologic process is believed to start with an acute myocarditic phase (probably viral in most cases), followed by a variable latent phase, then a phase of chronic fibrosis and diffuse loss of myocardial myocytes due to an autoimmune reaction to virus-altered myocytes. Regardless of the cause, the result is dilation, thinning, and compensatory hypertrophy of the remaining myocardium interspersed with fibrosis. Altered ventricular geometry often leads to secondary functional mitral or tricuspid regurgitation and atrial dilation. The primary result is impaired ventricular systolic function reflected by a low ejection fraction (EF). Cardiac output is maintained through tachycardia and an increased end-diastolic volume, which increases wall tension and myocardial O2 demand. Diastolic compliance and pressure become abnormal only late in the disease.
Diagnosis
Diagnosis depends on the characteristic history and physical examination and exclusion of other causes of ventricular failure (eg, systemic hypertension, primary valvular disease, MI). The ECG may show sinus tachycardia, low-voltage QRS, and nonspecific ST segment depression with low-voltage or inverted T waves. Sometimes pathologic Q waves are present in the precordial leads, simulating remote MI. Left bundle branch block is common. In about 25% of cases, differentiation from previous MI may be further complicated by chest pain, which may mimic angina pectoris but most often is atypical in character and position and not clearly related to exertion.
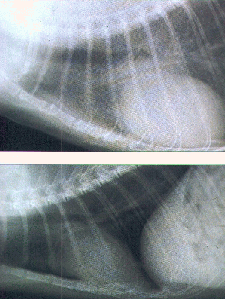
Chest x-ray reveals cardiomegaly that usually involves all chambers. Pleural effusion, particularly on the right, often accompanies raised pulmonary venous pressure and interstitial edema. M-mode and two-dimensional echocardiography shows dilated, hypokinetic cardiac chambers with reduced fractional shortening and rules out primary valvular disease or segmental wall motion abnormalities, as seen in discrete MI. Echocardiography also may reveal a mural thrombus, which frequently complicates dilated congestive cardiomyopathy. Radionuclide studies show diffusely dilated, hypokinetic cardiac chambers. In acute myocarditis, gallium scanning may identify an acute inflammatory phase (see Radionuclide Imaging in Ch. 198), whereas MRI reveals abnormal myocardial tissue texture.
Cardiac catheterization is reserved for patients in whom the diagnosis is in doubt after noninvasive investigation, particularly when chest pain is a symptom. Cardiac output can be normal or low, but EF is depressed and diffuse hypokinesis is seen on angiography. Valvular gradients and calcification are absent, and the coronary arteries are normal. Left ventricular end-diastolic pressure is elevated late in disease. Myocardial biopsy of either ventricle can be performed during catheterization. When special studies show a disproportionately low cardiac output in other forms of heart disease, the possibility of a coexisting cardiomyopathy should be considered.
Prognosis
The prognosis generally is poor: 70% of patients die in < 5 yr. Fifty percent of deaths are sudden, suggesting malignant arrhythmia. Unless a treatable primary cause can be found and eliminated (eg, alcohol, an infective agent), no specific therapy can prolong life. Prognosis is better if reactive hypertrophy is adequate to preserve ventricular wall thickness and is worse if thinning of ventricular walls is marked. Poor prognosis is related to poor ventricular function or frequent ventricular arrhythmia on 24-h ECG monitoring. Males survive half as long as females. Blacks survive half as long as whites.
Treatment
Treatment is specific for any underlying cause (eg, toxoplasmosis, thyrotoxicosis, beriberi) and may include elimination of potential toxins or myocardial depressants, therapy for low cardiac output and heart failure, and treatment of complications. However, most often, no cause is found. If possible, alcohol, some psychotherapeutic drugs, and electrolyte disorders should be eliminated. Therapy for heart failure and low cardiac output depends on the appropriate balance of afterload reduction, inotropic drugs, and preload reduction to optimize cardiac output and relieve systemic and pulmonary venous congestion.
Combined afterload and preload reduction with ACE inhibitors (eg, captopril, enalapril, lisinopril) or hydralazine plus nitrate (eg, isosorbide dinitrate) is the mainstay of therapy. These drugs favorably alter prognosis. Carvedilol and possibly other  -blockers prolong life and reduce morbidity. Digitalis glycosides, which reduce morbidity, are of value as weak inotropic drugs and for controlling the ventricular rate in patients with atrial fibrillation. Diuretics can lower left and right ventricular filling pressures so that pulmonary edema or significant hepatic congestion is avoided. Use of a phosphodiesterase inhibitor (eg, amrinone, milrinone [unavailable in USA]) or intermittent short-term (48- to 72-h) catecholamine infusion with dopamine or dobutamine is undergoing trials and temporarily helps some patients. These therapies have not been shown to prolong life. Corticosteroids with or without azathioprine and equine antithymocyte globulin may shorten the acute phase of certain inflammatory myocarditic myopathies proved by biopsy (eg, acute postviral or sarcoid myocarditis) but do not alter the course of chronic myopathy and are no longer used. Therefore, proof of active myocarditis on biopsy is recommended before starting corticosteroids or azathioprine.
-blockers prolong life and reduce morbidity. Digitalis glycosides, which reduce morbidity, are of value as weak inotropic drugs and for controlling the ventricular rate in patients with atrial fibrillation. Diuretics can lower left and right ventricular filling pressures so that pulmonary edema or significant hepatic congestion is avoided. Use of a phosphodiesterase inhibitor (eg, amrinone, milrinone [unavailable in USA]) or intermittent short-term (48- to 72-h) catecholamine infusion with dopamine or dobutamine is undergoing trials and temporarily helps some patients. These therapies have not been shown to prolong life. Corticosteroids with or without azathioprine and equine antithymocyte globulin may shorten the acute phase of certain inflammatory myocarditic myopathies proved by biopsy (eg, acute postviral or sarcoid myocarditis) but do not alter the course of chronic myopathy and are no longer used. Therefore, proof of active myocarditis on biopsy is recommended before starting corticosteroids or azathioprine.
Because of the risk of mural thrombus formation, prophylactic oral anticoagulant therapy helps prevent systemic or pulmonary emboli (see Ch. 72). Cardiac arrhythmias, which often complicate acute myocarditic phase myopathy and late chronic dilated phase, are treated with antiarrhythmic drugs as required (see Ch. 205). Caution: Most antiarrhythmics have a depressant effect on myocardial contractility; thus, potent negative inotropic drugs (eg, disopyramide, procainamide) are best avoided. The proarrhythmic effect of class I antiarrhythmic drugs (eg, encainide, flecainide) is greater the worse the ventricular function. Permanent pacemakers may be required if heart block complicates the chronic dilated phase; however, atrioventricular block during acute myocarditis often resolves, and permanent pacing usually is not needed.
In patients with underlying diffuse coronary artery disease with diffuse ischemic myopathy, therapy for angina pectoris with nitrates, -blockers, and Ca blockers may be indicated (see Ch. 201). However, the benefit of Ca blockers in controlling angina must be weighed against the negative inotropic effects; Ca blockers, except for amlodipine or felodipine, are best avoided. Some studies suggest that low-dose -blockers may be of benefit for some patients in whom a significant compensatory adrenergic response is causing chronic down-regulation of cardiac myocyte -adrenoceptors. Therapy must be started with very low doses (eg, carvedilol 6.25 mg or metoprolol 5 mg bid), and thorough evaluation for worsening of heart failure is essential. If tolerated, the dose is increased to 25 mg bid and 50 mg bid, respectively. Greatest benefit occurs with younger patients with dilated congestive cardiomyopathy of short duration. Females benefit more than males. Short-acting nifedipine normally acts as a vasodilator and afterload reducer; however, in the presence of cardiac decompensation, the reflex sympathetic response to arteriolar vasodilation may already be maximal, and the direct negative inotropic effect of the drug may manifest with a worsening of heart failure.
Appropriate rest, sleep, and stress avoidance are important, but prolonged bed rest should be prescribed only as symptoms dictate. Physical exercise within the limits imposed by symptoms improves overall well-being and may slightly prolong life.
Because of the grim prognosis, these patients represent the greatest proportion of heart transplantation recipients. Patients selected should be without associated systemic disease, psychologic disorders, or high, irreversible pulmonary vascular resistance, and they should generally be < 60 yr old because younger patients are the preferred recipients of these scarce organ resources.
A surgical procedure involving removal of strips of myocardium to remodel the dilated ventricle has shown promise in uncontrolled trials. Controlled trials comparing this procedure with optimal medical therapy are underway. A procedure whereby the latissimus dorsi muscle is wrapped around the failing ventricle and stimulated by a skeletal muscle pacemaker has been shown to be of no value.
Several ventricular assist devices having an internal power source with or without an external power source are being used to keep the patient alive pending heart transplantation or as long-term therapy instead of transplantation.
HYPERTROPHIC CARDIOMYOPATHY
Congenital or acquired disorders characterized by marked ventricular hypertrophy with diastolic dysfunction in the absence of an afterload demand (eg, valvular aortic stenosis, coarctation of the aorta, systemic hypertension).
Identifiable causes are listed in Table 203-2.
Pathology and Pathophysiology
The cardiac muscle is abnormal with cellular and myofibrillar disarray, although this finding is not specific to hypertrophic cardiomyopathy. Usually, the interventricular septum is hypertrophied more than the left ventricular posterior wall (asymmetric septal hypertrophy). In the most common asymmetric form of hypertrophic cardiomyopathy, there is marked hypertrophy and thickening of the upper interventricular septum below the aortic valve. During systole, the septum thickens and the anterior leaflet of the mitral valve, already abnormally oriented due to the abnormal shape of the ventricle, is sucked toward the septum, producing outflow tract obstruction. This is termed hypertrophic obstructive cardiomyopathy or asymmetric hypertrophic septal stenosis. It further reduces cardiac output, which is already abnormally low because of diastolic dysfunction caused by the noncompliant, hypertrophied ventricle.
Congenital hypertrophy is autosomally dominant in cases of asymmetric septal hypertrophy but not in other varieties. The most common abnormality is a missense point mutation in exon 13 of the cardiac myosin heavy chain gene on the DNA locus of chromosome 14. Less often, an abnormal  / cardiac myosin heavy chain hybrid gene is present. Other gene defects can also cause the disorder.
/ cardiac myosin heavy chain hybrid gene is present. Other gene defects can also cause the disorder.
The major consequence of hypertrophy is that the stiff, noncompliant chamber (usually the left ventricle) resists diastolic filling, leading to elevated end-diastolic pressure, which raises pulmonary venous pressure. Angina pectoris results from an imbalance between O2 demand by the hypertrophied myocardium and O2 supply via the coronary arteries, which may be compromised by the noncompliant myocardium. Inadequate capillary density relative to myocyte size as well as hyperplasia and hypertrophy of the intima and media of intramyocardial coronary arteries, which compromise lumen diameter, also produces ischemia in hypertrophic cardiomyopathy in the absence of epicardial coronary artery disease.
Effort-induced light-headedness and syncope are caused by inadequate cardiac output, sometimes worsened by an outflow tract gradient in those with asymmetric septal hypertrophy. The fall in cardiac output results from the decreased diastolic filling period related to effort-induced sinus tachycardia. Shortening of the diastolic filling period in the noncompliant, hypertrophied ventricle reduces preload and leads to increased apposition of the anterior leaflet of the mitral valve against the hypertrophied ventricular septum. Exercise also lowers peripheral vascular resistance and hence aortic root diastolic pressure. This may induce ischemia, which may lead to nonsustained ventricular or atrial arrhythmia, causing syncope. Syncope in hypertrophic cardiomyopathy is a clinical marker of increased probability of sudden death, which is believed to result from ventricular tachycardia or fibrillation.
Infective endocarditis can complicate hypertrophic cardiomyopathy because of mitral valve abnormality that appears to result from the altered ventricular geometry with anterior positioning of the papillary muscles and mitral apparatus and the Venturi effect produced by the rapid early systolic flow through the outflow tract. Heart block is sometimes a late complication. Midventricular hypertrophy leads to an intracavitary gradient at the papillary muscle level. The distal left ventricle may ultimately thin and dilate in an aneurysmal manner.
Symptoms, Signs, and Diagnosis
Clinical manifestations may occur alone or in any combination: Chest pain is usually typical angina related to exertion. Syncope is usually exertional and due to a combination of ischemia, arrhythmia, outflow tract obstruction, and poor diastolic filling of the ventricle. Dyspnea on exertion results from poor diastolic compliance of the left ventricle, which leads to a rapid rise in left ventricular end-diastolic pressure as flow increases. Outflow tract obstruction, by lowering cardiac output, may contribute to the dyspnea. Systolic function is preserved, and fatigability is seldom a complaint. Palpitations are produced by ventricular or atrial arrhythmias. Thus, symptoms of hypertrophic cardiomyopathy may simulate those of aortic stenosis or coronary artery disease.
Physical examination usually clarifies the differential diagnosis. Signs of raised venous pressure (eg, jugular venous distention, ascites, ankle edema, pleural effusion) are rare until the terminal phase. BP and heart rate are usually normal. The carotid pulse in cases with asymmetric septal hypertrophy and outflow tract obstruction has a brisk upstroke, a bifid peak due to dynamic obstruction in the latter part of systole, and a rapid downstroke. Precordial palpation reveals the apex beat in its normal position with a sustained thrust due to left ventricular hypertrophy. Sometimes a biphasic apical thrust can be appreciated in cases with severe outflow obstruction.
Systolic murmurs are usually present, but patients with apical and symmetric hypertrophic cardiomyopathy may have no murmur. Most common is a crescendo-diminuendo ejection-type murmur that does not radiate to the neck; it is best heard at the left sternal edge in the 3rd or 4th intercostal space. This murmur is caused by obstruction of left ventricular ejection (produced in systole when the hypertrophied interventricular septum and the anterior leaflet of the mitral valve approach each other). A mitral regurgitation murmur due to distortion of the mitral apparatus is heard in some patients. It has a characteristic blowing quality and is best heard at the apex, radiating toward the left axilla. Rarely, early or midsystolic clicks are heard. In some patients with right ventricular outflow tract narrowing, a systolic ejection murmur is heard in the second interspace at the left sternal border. An S4, almost always present, indicates a forceful atrial contraction against a poorly compliant left ventricle in late diastole (hear Audio 203-2).
The ejection murmur of hypertrophic cardiomyopathy can be altered by maneuvers to decrease venous return, reducing left ventricular diastolic volume and increasing apposition of the anterior mitral valve leaflet with the hypertrophied interventricular septum. Thus, Valsalva maneuver increases the intensity of the murmur (hear Audio 203-3), as will maneuvers to lower aortic pressure (eg, amyl nitrite inhalation) or a postextrasystolic contraction, by increasing the outflow tract pressure gradient. Handgrip raises aortic pressure, thereby reducing the murmur's intensity (hear Audio 203-4).
Laboratory Findings
Noninvasive tests to confirm the diagnosis have generally replaced heart catheterization. The ECG usually shows voltage criteria for left ventricular hypertrophy. Asymmetric septal hypertrophy is often suggested by very deep septal Q waves in leads I, aVL, V5, and V6; a QS complex sometimes occurs in V1 and V2, simulating previous septal infarction. The T waves are abnormal in most cases; the most common finding is deep symmetric T-wave inversion in leads I, aVL, V5, and V6. ST segment depression in the same leads is common. The P wave is often broad and notched in leads II, III, and aVF, with a biphasic P wave in V1 and V2, indicative of left atrial hypertrophy. Preexcitation phenomenon of the Wolff-Parkinson-White syndrome type occurs more often than by chance alone and is one of the mechanisms of arrhythmia-induced palpitations.
The chest x-ray is often deceptively normal looking because hypertrophy occurs at the expense of the ventricular cavities; a globular left ventricular contour within a normal-sized cardiac silhouette may be the only abnormality. Cardiac fluoroscopy will rule out aortic valve calcification.
M-mode and two-dimensional echocardiography with Doppler study is the best noninvasive diagnostic technique. Thickened ventricular walls can be measured, allowing forms of hypertrophic cardiomyopathy to be differentiated (see Fig. 203-3). Anterior positioning of the papillary muscles and mitral apparatus is usual. Outflow tract obstruction can often be quantitated by observing the degree of systolic anterior movement of the anterior leaflet of the mitral valve and its degree and duration of apposition to the hypertrophied interventricular septum. Doppler velocity analysis of flow across the ventricular outflow tract can quantitate the gradient and area of the stenotic segment and is particularly useful for monitoring the effect of medical or surgical treatment. Doppler analysis of mitral inflow velocity in diastole usually shows evidence of diastolic dysfunction of the left ventricle; left ventricular fractional shortening and ejection fraction (EF) are normal or increased. Midsystolic closure of the aortic valve sometimes occurs in patients with severe outflow tract obstruction. Radionuclide angiography shows a small ventricular cavity with a normal or high EF.
Cardiac catheterization is usually performed only when surgical therapy is considered. Intraventricular pressure gradients may be found in the left and, less commonly, the right ventricle. The gradient rises in a postextrasystolic beat, during Valsalva maneuver, and after amyl nitrite inhalation. End-diastolic pressure is often high because of poor ventricular compliance. EF is normal or high. Ventriculography shows characteristic chamber deformity depending on the form of hypertrophic cardiomyopathy and sometimes confirms mitral valve regurgitation. The coronary arteries are usually widely patent with torrential flow, although sophisticated metabolic studies may reveal myocardial ischemia due to intramyocardial artery lumen reduction, capillary/myocyte imbalance, and abnormal ventricular wall stress. In older patients, coronary artery disease may coexist.
A few cases gradually lose myocytes, probably from chronic diffuse ischemia as a result of capillary/myocyte imbalance. As myocytes die, they are replaced by diffuse fibrosis, and the hypertrophied ventricle with diastolic dysfunction gradually becomes dilated with systolic dysfunction and becomes an end-stage congestive cardiomyopathy.
Prognosis
Prognosis is guarded; the annual mortality rate is 4%. (The mortality rate is inversely proportional to the age at which symptoms appear and greatest in those with frequent nonsustained ventricular tachycardia, syncope, or resuscitated sudden death.) Family history of sudden death in young patients and angina or effort dyspnea in patients > 45 yr denote a worse prognosis. Sudden death is most common, with chronic heart failure occurring less often. Genetic counseling is appropriate for patients with asymmetric septal hypertrophy, which appears to accelerate during puberty.
Treatment
Therapy is directed primarily at abnormal diastolic compliance. -Adrenoceptor blockers and Ca blockers alone or in combination are the mainstays of treatment. Both decrease myocardial contractility, which dilates the heart and decreases outflow obstruction, improving diastolic ventricular function. -Blockers and rate-limiting Ca blockers also slow the heart rate, prolonging the diastolic filling period and thus decreasing outflow obstruction. -Blockers with intrinsic sympathomimetic action (eg, pindolol, oxprenolol, acebutolol) are best avoided. Ca blockers vary in their negative inotropic effect and arterial vasodilator capacity. It is important to choose a weak vasodilator that has a significant depressant effect on contractility. Verapamil is the Ca blocker of choice for hypertrophic cardiomyopathy.
Drugs that reduce preload (eg, nitrates, diuretics, ACE inhibitors, angiotensin blockers) decrease chamber size and make the signs and symptoms worse. Inotropic drugs (eg, digitalis glycosides, catecholamines) worsen outflow tract obstruction, do not relieve the high end-diastolic pressure, and may induce arrhythmias. Vasodilators increase the outflow tract gradient and produce a reflex tachycardia that further decreases ventricular diastolic function. Although antiarrhythmic therapy can be prescribed for arrhythmias proved by ECG or 24-h ambulatory monitoring, there is no evidence that it alters the risk of sudden death. However, uncontrolled retrospective studies of amiodarone suggest it may reduce mortality in patients with nonsustained ventricular tachyarrhythmias or syncope. The antifibrillatory action of -blockers may help to prevent sudden death, but this has not been proved. Disopyramide has a negative inotropic effect and has been used as an antiarrhythmic and as a negative inotropic drug.
Defibrillators have been implanted in persons who have been resuscitated from sudden death. Although this treatment is reasonable, it has not been proven to reduce overall mortality in hypertrophic cardiomyopathy. Antibiotic prophylaxis for infective endocarditis should be recommended (see Ch. 208). Competitive sports should be avoided because many sudden deaths occur with increased exertion.
Patients who progress to the dilated congestive phase of the disease are managed in the same manner as those with dilated cardiomyopathy with predominant systolic dysfunction.
Septal myotomy or myomectomy is reserved for patients with incapacitating symptoms despite medical therapy, in whom outflow tract obstruction has been shown by echocardiography and catheterization studies. It lessens symptoms in most carefully selected cases but does not alter mortality. Selective septal infarction using absolute ethanol injection through steerable catheters inserted in septal perforator branches of the anterior descending artery has shown promise and may be an alternative to septal myectomy. In a few cases, mitral valve tailoring or replacement has been done for severe mitral dysfunction; this coincidentally eliminates the outflow tract gradient. Dual chamber pacemakers have been used to alter the sequence of ventricular depolarization in some patients with outflow tract obstruction. In most cases, the severity of obstruction was reduced and symptoms lessened. The long-term effect of this treatment and the effect on mortality require further study.
RESTRICTIVE CARDIOMYOPATHY
Myocardial disorders characterized by rigid, noncompliant ventricular walls that resist diastolic filling of one or both ventricles, most commonly the left.
This form of cardiomyopathy is the least prevalent.
Etiology and Pathology
The cause is usually unknown (for identified causes, see Table 203-2). Amyloidosis involving the myocardium is usually systemic, as is iron infiltration in hemochromatosis. Sarcoidosis and Fabry's disease involve the myocardium, and nodal conduction tissue can be involved. Löffler's disease (a subcategory of hypereosinophilic syndrome with primary cardiac involvement) is a cause of restrictive cardiomyopathy. It occurs in the tropics. It begins as an acute arteritis with eosinophilia, with subsequent thrombus formation on the endocardium, chordae, and atrioventricular valves, progressing to fibrosis. Endocardial fibrosis occurs in temperate zones and involves only the left ventricle.
Restrictive cardiomyopathy is divided into a diffuse nonobliterative variety in which the myocardium is infiltrated by abnormal substance (eg, amyloidosis) and an obliterative variety in which the endocardium and subendocardium are fibrosed (eg, endomyocardial fibrosis). Either may be nondiffuse if the disease affects only one chamber or part of one chamber unevenly.
Pathophysiology
The pathophysiologic consequences include endocardial thickening or myocardial infiltration with loss of myocytes, compensatory hypertrophy, and fibrosis. Any of these may lead to atrioventricular valve malfunction resulting in mitral or tricuspid regurgitation. Involvement of nodal and conduction tissue results in sinoatrial node dysfunction and, in some cases, various grades of heart block. Amyloidosis can involve the coronary arteries.
The main hemodynamic consequence of these pathologic states is diastolic dysfunction with a rigid, noncompliant chamber with a high filling pressure. Systolic function may deteriorate if compensatory hypertrophy is inadequate in cases of infiltrated or fibrosed chambers. Mural thrombosis and systemic emboli can complicate the restrictive or obliterative variety.
Symptoms, Signs, and Diagnosis
Similar to hypertrophic cardiomyopathy, the main dysfunction is abnormal compliance and diastolic filling of one or both ventricles, most commonly the left. Symptoms are due to high diastolic pressure, giving rise to pulmonary venous hypertension with effort dyspnea and orthopnea and peripheral edema when the right ventricle is involved. Limited effort is a consequence of a fixed cardiac output due to resistance to ventricular filling. Angina and syncope are uncommon, but atrial and ventricular arrhythmias and heart block are common.
Physical examination reveals a quiet precordium, a low volume and rapid carotid pulse, pulmonary rales, and pronounced neck vein distention with a rapid y descent (see Fig. 197-1). An S4 is heard in virtually all cases, and an S3 may occur and must be differentiated from a precordial knock, but often no murmur is present. In some cases, a murmur of functional mitral or tricuspid regurgitation results from changes in chordae or ventricular geometry as a result of infiltration or fibrosis of myocardium and endocardium. Symptoms and signs thus closely mimic constrictive pericarditis; noninvasive tests--including CT, which demonstrates a normal pericardium--may help make this differentiation, but occasionally even heart catheterization is not diagnostic, and thoracotomy is required to explore the pericardium.
The ECG is usually nonspecifically abnormal, showing ST segment and T-wave abnormalities and sometimes low voltage. Pathologic Q waves sometimes occur without previous MI. Left ventricular hypertrophy due to compensatory hypertrophy of the myocardium sometimes occurs. On chest x-ray, the heart size is often normal or small but can be enlarged in late-stage amyloidosis or hemochromatosis.
Echocardiography shows normal systolic function. The atria are often dilated. Disease due to amyloidosis shows an unusually bright echo pattern from the myocardium. Echocardiography helps differentiate constrictive pericarditis with its thickened pericardium, but paradoxical septal motion can occur in either disorder. Myocardial hypertrophy often occurs in restrictive myopathy. MRI reveals abnormal myocardial texture in diseases with myocardial infiltration (eg, of amyloid or iron).
Cardiac catheterization and myocardial biopsy are often necessary. High atrial pressure with a prominent y descent and an early diastolic dip followed by a high diastolic plateau in the ventricular pressure curve are found. Unlike in constrictive pericarditis, diastolic pressure usually is a few millimeters of mercury higher in the left ventricle than in the right. Angiography reveals normal-sized ventricular cavities with normal or decreased systolic shortening. Functional atrioventricular valve regurgitation may result from infiltration of myocardium and papillary muscles or endocardial thickening. Biopsy can demonstrate endocardial fibrosis and thickening, myocardial infiltration with iron or amyloid, or chronic myocardial fibrosis. Coronary angiography is normal, except in rare cases of amyloidosis involving the epicardial coronary arteries.
Primary causes of restrictive cardiomyopathy should be sought (eg, rectal biopsy for amyloidosis, iron studies or biopsy for hemochromatosis).
Prognosis and Treatment
Prognosis is poor (see Table 203-3), similar to that of dilated congestive cardiomyopathy (see above).
No therapy is available for most patients. Diuretics must be used with caution because they can lower preload, on which the noncompliant ventricles depend to maintain cardiac output. Digitalis does little to alter the hemodynamic abnormality and may be dangerous in amyloidosis cardiomyopathy, in which extreme digitalis sensitivity is common. Afterload reducers may induce profound hypotension and usually are not of value.
Hemochromatosis may improve with regular phlebotomy to reduce the body's iron stores, and patients with active biopsy-proven sarcoidosis will respond to corticosteroids. Patients in the acute phase of the hypereosinophilic syndrome may respond to corticosteroids and cytotoxic drugs (eg, hydroxyurea). Rarely, in the chronic phase, patients with endocardial fibroelastosis or Löffler's disease improve after surgical debridement of the endocardial fibrotic and thrombotic thickening and freeing up of chordae and valve tissue. Sometimes atrioventricular valve replacement has helped severe functional atrioventricular valve regurgitation. In some cases of significant compensatory hypertrophy, Ca blockers might be of value. Hemodynamic monitoring during initiation of such therapy to confirm its efficacy is prudent.